The azide group (-N₃) has emerged as one of the most versatile chemical handles in peptide chemistry, primarily due to its central role in click chemistry reactions. This small, bioorthogonal functional group enables the selective conjugation of peptides to a vast array of molecules, including fluorophores, drugs, oligonucleotides, and surfaces, under mild, physiologically compatible conditions. However, the decision to incorporate an azide into a peptide sequence requires careful consideration of the intended application, the availability of compatible conjugation partners, and the potential impact on peptide synthesis and stability. This article provides a framework for evaluating whether an azide modification aligns with your research objectives.
Key Takeaways
- An azide group serves as a bioorthogonal handle for click chemistry, enabling specific conjugation without interfering with native biological functions.
- Two primary click chemistry pathways exist: copper-catalyzed azide-alkyne cycloaddition (CuAAC) and copper-free strain-promoted azide-alkyne cycloaddition (SPAAC) using DBCO or related cyclooctynes.
- Copper-free methods are strongly preferred for biological applications involving live cells, proteins, or sensitive biomolecules, as copper ions can cause denaturation and toxicity.
- Azide incorporation is most commonly achieved at the N-terminus or on lysine side chains using commercially available Fmoc-protected azido-amino acid building blocks.
- Alternative bioorthogonal strategies, such as methyltetrazine-trans-cyclooctene (TCO) IEDDA reactions, may offer faster kinetics for certain applications.
Introduction to the Azide Functional Group in Peptides
What Makes the Azide Group Unique?
The azide moiety is a small, linear functional group consisting of three nitrogen atoms in a chain. Its value in peptide chemistry stems from three key properties:
- Bioorthogonality: Azides are absent from native biological systems, meaning they do not react with endogenous functional groups such as amines, thiols, or carboxylates.
- Selective Reactivity: Azides undergo highly specific cycloaddition reactions with alkynes, particularly strained cyclooctynes, forming stable 1,2,3-triazole linkages.
- Small Size: Minimal steric footprint reduces the likelihood of interfering with peptide folding, receptor binding, or biological activity.
The Click Chemistry Revolution
The discovery that azides and alkynes could be linked under mild conditions, coined “click chemistry” and recognized with the 2022 Nobel Prize in Chemistry, has transformed bioconjugation. Two principal approaches dominate peptide applications:
- CuAAC (Copper-Catalyzed Azide-Alkyne Cycloaddition): Utilizes a copper(I) catalyst to drive the reaction between an azide and a terminal alkyne. While highly efficient, the copper catalyst can damage sensitive biomolecules and is toxic to living systems.
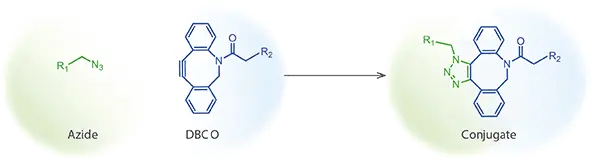
- SPAAC (Strain-Promoted Azide-Alkyne Cycloaddition): Employs strained cyclooctynes such as DBCO (dibenzocyclooctyne) or BCN, eliminating the need for a copper catalyst. This “copper-free click chemistry” proceeds under physiological conditions and is compatible with live cells and organisms.
Find out more about peptide synthesis here.
Decision Framework: When to Include an Azide Group
Consideration 1: Your Primary Application
For Bioconjugation to Complex Biomolecules
If your goal is to conjugate a peptide to an antibody, protein, or oligonucleotide, including an azide is an excellent strategy. The standard workflow involves:
- Activating the target molecule (e.g., antibody) with a DBCO-NHS ester, which attaches DBCO groups to surface lysines.
- Reacting the DBCO-functionalized target with your azide-modified peptide under mild conditions (PBS, 4°C to room temperature, overnight).
This approach yields stable triazole-linked conjugates without the need for toxic catalysts. It has been successfully applied to antibody-DNA conjugates, antibody-drug conjugates, and antibody-peptide systems.
For Fluorescent Labeling and Imaging
Azide-modified peptides can be conjugated to alkyne- or DBCO-functionalized fluorophores for cellular imaging applications. Recent advances have demonstrated rapid and quantitative labeling of azide-containing peptides using copper-chelating azide designs that accelerate CuAAC reactions by up to three orders of magnitude compared to traditional catalysts.
For Peptide-Peptide Ligation and Cyclization
Azide groups enable the joining of peptide fragments or the formation of cyclic peptides. By incorporating an azide on one fragment and an alkyne on another, a triazole bridge can be formed that mimics an amide bond while conferring resistance to enzymatic degradation. This approach has been used to create stapled peptides with enhanced helical content and stability.
Consideration 2: Available Click Chemistry Partners
Before committing to an azide modification, verify that you have access to a compatible alkyne- or DBCO-functionalized conjugation partner. Common options include:
Consideration 3: Alternative Bioorthogonal Handles
While azides are powerful, they are not the only option. Methyltetrazine-maleimide linkers offer an alternative bioorthogonal strategy via inverse electron-demand Diels-Alder (IEDDA) reactions with TCO or norbornene partners. These reactions are among the fastest bioorthogonal ligations known, completing in minutes rather than hours. The choice between azide and tetrazine often depends on:
- Kinetic requirements: IEDDA is significantly faster than SPAAC.
- Partner availability: Azide-DBCO pairs are more widely commercially available.
- Application context: Live-cell labeling may favor one over the other based on background reactivity.
Synthesis and Incorporation Strategies
Where to Place the Azide Group
Azides can be incorporated at multiple positions within a peptide sequence:
- N-terminus: Using azido-acetic acid or Fmoc-protected azido-amine building blocks during solid-phase peptide synthesis (SPPS).
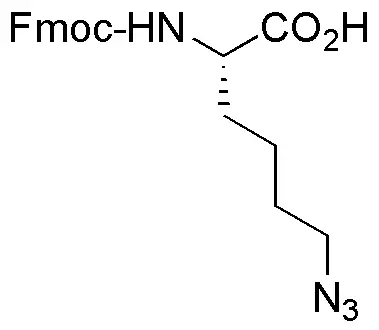
- Lysine side chains: Using commercially available Fmoc-Lys(N₃)-OH or Fmoc-Lys(azide)-OH building blocks.
- C-terminus: Achieved through specialized resins or solution-phase modification.
Protecting Group Considerations
When incorporating azide-functionalized amino acids, standard Fmoc/tBu SPPS is compatible, provided the azide group is stable to the acidic and basic conditions used in synthesis and cleavage. Fmoc-protected ω-azido-L-amino acids are commercially available for introducing azides at various chain lengths.
Purification and Stability
Azide-containing peptides should be purified by standard reverse-phase HPLC and stored desiccated at -20°C. The azide group is generally stable under these conditions but may be reduced to a primary amine by strong reducing agents such as dithiothreitol (DTT) or tris(2-carboxyethyl)phosphine (TCEP), a consideration if your peptide also contains disulfide bonds.
Find out about high-speed RUSH synthesis.
Frequently Asked Questions (FAQ)
What is the difference between CuAAC and SPAAC click chemistry?
CuAAC (copper-catalyzed azide-alkyne cycloaddition) requires a copper(I) catalyst to facilitate the reaction between an azide and a terminal alkyne. SPAAC (strain-promoted azide-alkyne cycloaddition) uses strained cyclooctynes such as DBCO and proceeds without a catalyst. For biological applications involving live cells, proteins, or sensitive biomolecules, copper-free SPAAC is strongly preferred to avoid copper-induced toxicity and denaturation.
Can I incorporate an azide into any peptide sequence?
Yes, azides can be incorporated at the N-terminus, on lysine side chains, or at the C-terminus using commercially available building blocks. However, careful design is needed if the peptide contains other reactive groups (e.g., disulfide bonds) that may interfere with subsequent click reactions.
Is the azide group stable during peptide synthesis?
Yes. The azide group is stable under standard Fmoc SPPS conditions, including exposure to piperidine for deprotection and TFA for cleavage. However, it may be reduced by strong reducing agents such as DTT or TCEP.
Sinozaki, Y., Otani, R., Tanimoto, H., & Tomohiro, T. (2025). Synthesis and Application of Azide‐Incorporated Copper‐Chelating Peptides for Efficient Click Conjugation Reaction. Journal of Heterocyclic Chemistry, 62(7), 419–429. https://doi.org/10.1002/jhet.4955
Rathinam, S., Sørensen, K. K., Hjálmarsdóttir, M. Á., Thygesen, M. B., & Másson, M. (2024). Conjugation of CRAMP18–35 Peptide to Chitosan and Hydroxypropyl Chitosan via Copper-Catalyzed Azide–Alkyne Cycloaddition and Investigation of Antibacterial Activity. International Journal of Molecular Sciences, 25(17), 9440. https://doi.org/10.3390/ijms25179440